5. Read mapping¶
5.1. Preface¶
In this section we will use our skill on the command-line interface to map our reads from the evolved line to our ancestral reference genome.
Note
You will encounter some To-do sections at times. Write the solutions and answers into a text-file.
5.2. Overview¶
The part of the workflow we will work on in this section can be viewed in Fig. 5.1.
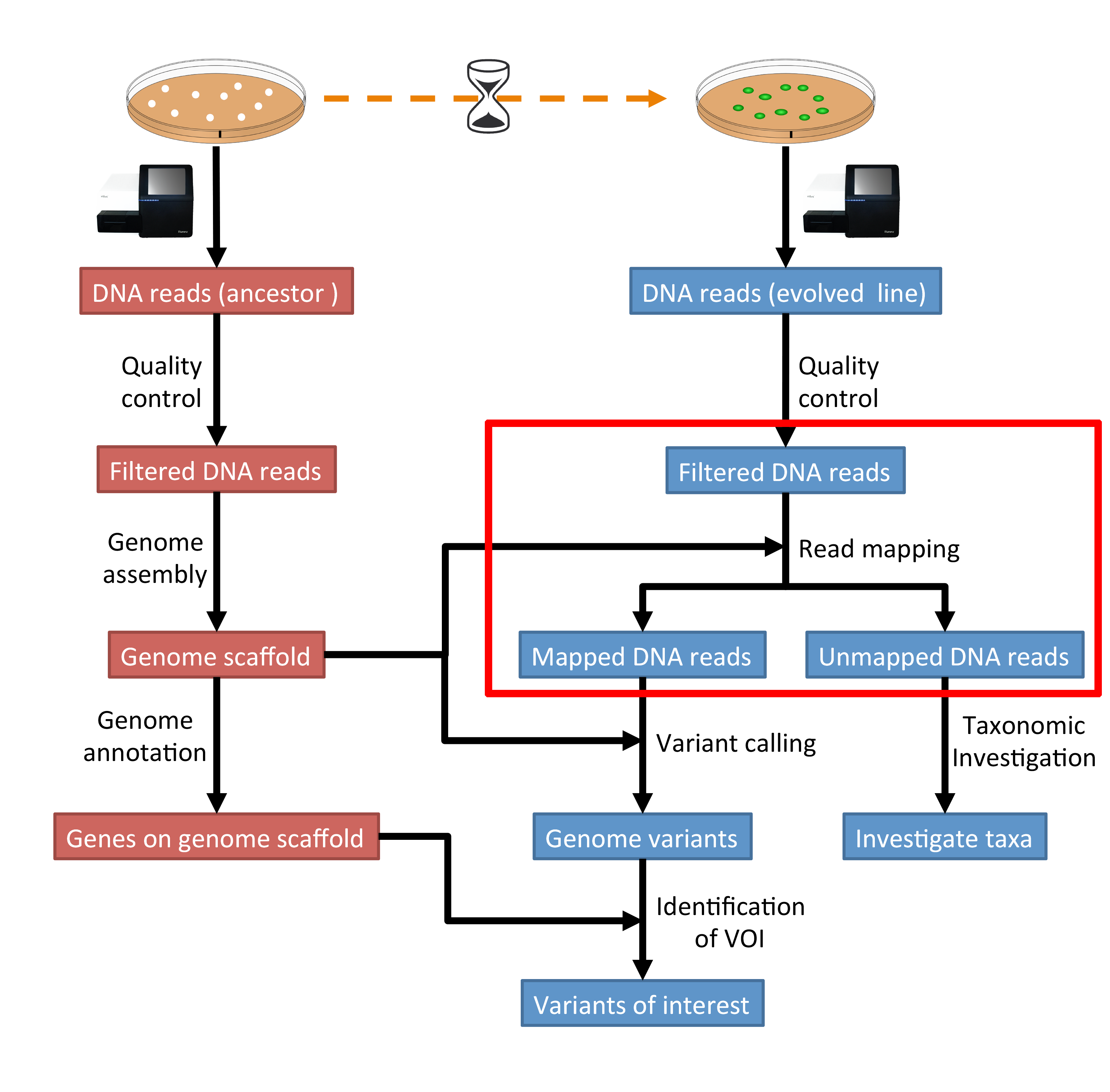
Fig. 5.1 The part of the workflow we will work on in this section marked in red.¶
5.3. Learning outcomes¶
After studying this section of the tutorial you should be able to:
Explain the process of sequence read mapping.
Use bioinformatics tools to map sequencing reads to a reference genome.
Filter mapped reads based on quality.
5.4. Before we start¶
Lets see how our directory structure looks so far:
$ cd ~/analysis
# create a mapping result directory
$ mkdir mappings
$ ls -1F
assembly/
data/
mappings/
multiqc_data/
multiqc_report.html
trimmed/
trimmed-fastqc/
Attention
If you have not run the previous sections on Quality control and Genome assembly, you can download the trimmed data and the genome assembly needed for this section here: Downloads. Download the files to the ~/analysis directory and decompress. Alternatively on the CLI try:
cd ~/analysis
wget -O trimmed.tar.gz https://osf.io/m3wpr/download
tar xvzf trimmed.tar.gz
wget -O assembly.tar.gz https://osf.io/t2zpm/download
tar xvzf assembly.tar.gz
5.5. Mapping sequence reads to a reference genome¶
We want to map the sequencing reads to the ancestral reference genome. We are going to use the quality trimmed forward and backward DNA sequences of the evolved line and use a program called BWA to map the reads.
Todo
Discuss briefly why we are using the ancestral genome as a reference genome as opposed to a genome for the evolved line.
5.5.1. Downloading the reference genome assembly¶
Todo
In the assembly section at “Genome assembly”, we created a genome assembly. However, we actually used sub-sampled data as otherwise the assemblies would have taken a long time to finish. To continue, please download the assembly created on the complete dataset (Downloads). Unarchive and uncompress the files with tar -xvzf assembly.tar.gz.
5.6. BWA¶
5.6.1. Overview¶
BWA is a short read aligner, that can take a reference genome and map single- or paired-end sequence data to it [LI2009]. It requires an indexing step in which one supplies the reference genome and BWA will create an index that in the subsequent steps will be used for aligning the reads to the reference genome. While this step can take some time, the good thing is the index can be reused over and over. The general command structure of the BWA tools we are going to use are shown below:
# bwa index help
$ bwa index
# indexing
$ bwa index path/to/reference-genome.fa
# bwa mem help
$ bwa mem
# single-end mapping, general command structure, adjust to your case
$ bwa mem path/to/reference-genome.fa path/to/reads.fq.gz > path/to/aln-se.sam
# paired-end mapping, general command structure, adjust to your case
$ bwa mem path/to/reference-genome.fa path/to/read1.fq.gz path/to/read2.fq.gz > path/to/aln-pe.sam
5.6.2. Creating a reference index for mapping¶
Todo
Create an BWA index for our reference genome assembly. Attention! Remember which file you need to submit to BWA.
Hint
Should you not get it right, try the commands in Code: BWA indexing.
5.6.3. Mapping reads in a paired-end manner¶
Now that we have created our index, it is time to map the trimmed sequencing reads of our two evolved line to the reference genome.
Todo
Use the correct bwa mem command structure from above and map the reads of the two evolved line to the reference genome.
Hint
Should you not get it right, try the commands in Code: BWA mapping.
5.7. The sam mapping file-format¶
BWA, like most mappers, will produce a mapping file in sam-format. Have a look into the sam-file that was created by either program. A quick overview of the sam-format can be found here and even more information can be found here. Briefly, first there are a lot of header lines. Then, for each read, that mapped to the reference, there is one line.
The columns of such a line in the mapping file are described in Table 5.1.
Col |
Field |
Description |
|---|---|---|
1 |
QNAME |
Query (pair) NAME |
2 |
FLAG |
bitwise FLAG |
3 |
RNAME |
Reference sequence NAME |
4 |
POS |
1-based leftmost POSition/coordinate of clipped sequence |
5 |
MAPQ |
MAPping Quality (Phred-scaled) |
6 |
CIAGR |
extended CIGAR string |
7 |
MRNM |
Mate Reference sequence NaMe (‘=’ if same as RNAME) |
8 |
MPOS |
1-based Mate POSition |
9 |
ISIZE |
Inferred insert SIZE |
10 |
SEQ |
query SEQuence on the same strand as the reference |
11 |
QUAL |
query QUALity (ASCII-33 gives the Phred base quality) |
12 |
OPT |
variable OPTional fields in the format TAG:VTYPE:VALUE |
One line of a mapped read can be seen here:
M02810:197:000000000-AV55U:1:1101:10000:11540 83 NODE_1_length_1419525_cov_15.3898 607378 60 151M = 607100 -429 TATGGTATCACTTATGGTATCACTTATGGCTATCACTAATGGCTATCACTTATGGTATCACTTATGACTATCAGACGTTATTACTATCAGACGATAACTATCAGACTTTATTACTATCACTTTCATATTACCCACTATCATCCCTTCTTTA FHGHHHHHGGGHHHHHHHHHHHHHHHHHHGHHHHHHHHHHHGHHHHHGHHHHHHHHGDHHHHHHHHGHHHHGHHHGHHHHHHFHHHHGHHHHIHHHHHHHHHHHHHHHHHHHGHHHHHGHGHHHHHHHHEGGGGGGGGGFBCFFFFCCCCC NM:i:0 MD:Z:151 AS:i:151 XS:i:0
It basically defines the read and the position within the reference genome, where the read mapped and a quality of the mapping.
5.8. Mapping post-processing¶
5.8.1. Fix mates and compress¶
Because aligners can sometimes leave unusual SAM flag information on SAM records, it is helpful when working with many tools to first clean up read pairing information and flags with SAMtools.
We are going to produce also compressed bam output for efficient storing of and access to the mapped reads.
Note, samtools fixmate expects name-sorted input files, which we can achieve with samtools sort -n.
$ samtools sort -n -O sam mappings/evol1.sam | samtools fixmate -m -O bam - mappings/evol1.fixmate.bam
-m: Add ms (mate score) tags. These are used by markdup (below) to select the best reads to keep.-O bam: specifies that we want compressed bam output from fixmate
Attention
The step of sam to bam-file conversion might take a few minutes to finish, depending on how big your mapping file is.
We will be using the SAM flag information later below to extract specific alignments.
Hint
A very useful tools to explain flags can be found here.
Once we have bam-file, we can also delete the original sam-file as it requires too much space and we can always recreate it from the bam-file.
$ rm mappings/evol1.sam
5.8.2. Sorting¶
We are going to use SAMtools again to sort the bam-file into coordinate order:
# convert to bam file and sort
$ samtools sort -O bam -o mappings/evol1.sorted.bam mappings/evol1.fixmate.bam
# Once it successfully finished, delete the fixmate file to save space
$ rm mappings/evol1.fixmate.bam
-o: specifies the name of the output file.-O bam: specifies that the output will be bam-format
5.8.3. Remove duplicates¶
In this step we remove duplicate reads. The main purpose of removing duplicates is to mitigate the effects of PCR amplification bias introduced during library construction. It should be noted that this step is not always recommended. It depends on the research question. In SNP calling it is a good idea to remove duplicates, as the statistics used in the tools that call SNPs sub-sequently expect this (most tools anyways). However, for other research questions that use mapping, you might not want to remove duplicates, e.g. RNA-seq.
$ samtools markdup -r -S mappings/evol1.sorted.bam mappings/evol1.sorted.dedup.bam
# if it worked, delete the original file
$ rm mappings/evol1.sorted.bam
Todo
Figure out what “PCR amplification bias” means.
Note
Should you be unable to do the post-processing steps, you can download the mapped data from Downloads.
5.9. Mapping statistics¶
5.9.1. Stats with SAMtools¶
Lets get an mapping overview:
$ samtools flagstat mappings/evol1.sorted.dedup.bam
Todo
Look at the mapping statistics and understand their meaning. Discuss your results. Explain why we may find mapped reads that have their mate mapped to a different chromosome/contig? Can they be used for something?
For the sorted bam-file we can get read depth for at all positions of the reference genome, e.g. how many reads are overlapping the genomic position.
$ samtools depth mappings/evol1.sorted.dedup.bam | gzip > mappings/evol1.depth.txt.gz
Todo
Extract the depth values for contig 20 and load the data into R, calculate some statistics of our scaffold.
$ zcat mappings/evol1.depth.txt.gz | egrep '^NODE_20_' | gzip > mappings/NODE_20.depth.txt.gz
Now we quickly use some R to make a coverage plot for contig NODE20.
Open a R shell by typing R on the command-line of the shell.
x <- read.table('mappings/NODE_20.depth.txt.gz', sep='\t', header=FALSE, strip.white=TRUE)
# Look at the beginning of x
head(x)
# calculate average depth
mean(x[,3])
# std dev
sqrt(var(x[,3]))
# mark areas that have a coverage below 20 in red
plot(x[,2], x[,3], col = ifelse(x[,3] < 20,'red','black'), pch=19, xlab='postion', ylab='coverage')
# to save a plot
png('mappings/covNODE20.png', width = 1200, height = 500)
plot(x[,2], x[,3], col = ifelse(x[,3] < 20,'red','black'), pch=19, xlab='postion', ylab='coverage')
dev.off()
The result plot will be looking similar to the one in Fig. 5.2
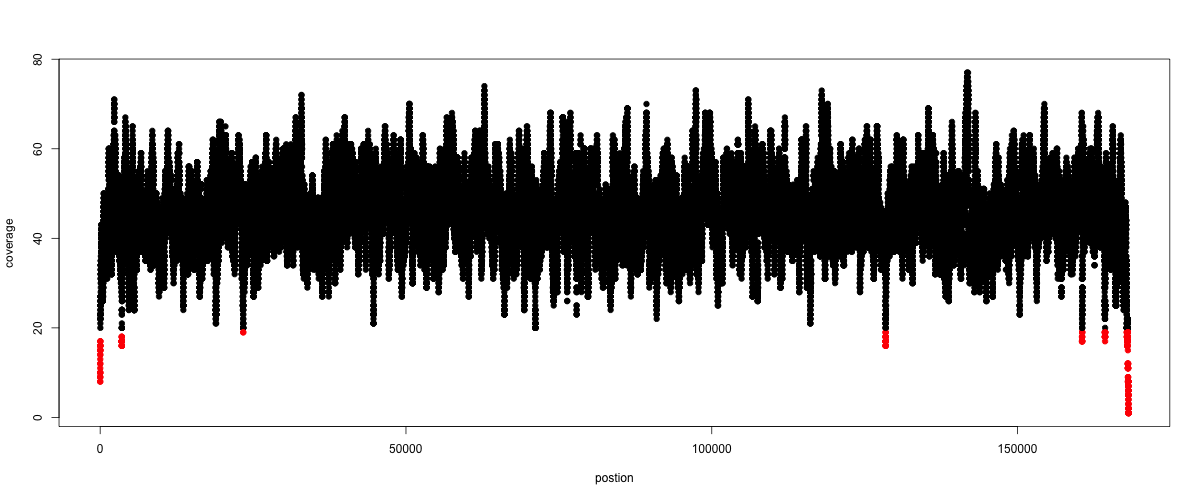
Fig. 5.2 A example coverage plot for a contig with highlighted in red regions with a coverage below 20 reads.¶
Todo
Look at the created plot. Explain why it makes sense that you find relatively bad coverage at the beginning and the end of the contig.
5.9.2. Stats with QualiMap¶
For a more in depth analysis of the mappings, one can use QualiMap [OKO2015].
QualiMap examines sequencing alignment data in SAM/BAM files according to the features of the mapped reads and provides an overall view of the data that helps to the detect biases in the sequencing and/or mapping of the data and eases decision-making for further analysis.
Run QualiMap with:
$ qualimap bamqc -bam mappings/evol1.sorted.dedup.bam
# Once finsished open reult page with
$ firefox mappings/evol1.sorted.dedup_stats/qualimapReport.html
This will create a report in the mapping folder. See this webpage to get help on the sections in the report.
Todo
Investigate the mapping of the evolved sample. Write down your observations.
5.10. Sub-selecting reads¶
It is important to remember that the mapping commands we used above, without additional parameters to sub-select specific alignments (e.g. for Bowtie2 there are options like --no-mixed, which suppresses unpaired alignments for paired reads or --no-discordant, which suppresses discordant alignments for paired reads, etc.), are going to output all reads, including unmapped reads, multi-mapping reads, unpaired reads, discordant read pairs, etc. in one file.
We can sub-select from the output reads we want to analyse further using SAMtools.
Todo
Explain what concordant and discordant read pairs are? Look at the Bowtie2 manual.
5.10.1. Concordant reads¶
We can select read-pair that have been mapped in a correct manner (same chromosome/contig, correct orientation to each other, distance between reads is not stupid).
Attention
We show the command here, but we are not going to use it.
$ samtools view -h -b -f 3 mappings/evol1.sorted.dedup.bam > mappings/evol1.sorted.dedup.concordant.bam
-b: Output will be bam-format-f 3: Only extract correctly paired reads.-fextracts alignments with the specified SAM flag set.
Todo
Our final aim is to identify variants. For a particular class of variants, it is not the best idea to only focus on concordant reads. Why is that?
5.10.2. Quality-based sub-selection¶
In this section we want to sub-select reads based on the quality of the mapping. It seems a reasonable idea to only keep good mapping reads. As the SAM-format contains at column 5 the \(MAPQ\) value, which we established earlier is the “MAPping Quality” in Phred-scaled, this seems easily achieved. The formula to calculate the \(MAPQ\) value is: \(MAPQ=-10*log10(p)\), where \(p\) is the probability that the read is mapped wrongly. However, there is a problem! While the MAPQ information would be very helpful indeed, the way that various tools implement this value differs. A good overview can be found here. Bottom-line is that we need to be aware that different tools use this value in different ways and the it is good to know the information that is encoded in the value. Once you dig deeper into the mechanics of the \(MAPQ\) implementation it becomes clear that this is not an easy topic. If you want to know more about the \(MAPQ\) topic, please follow the link above.
For the sake of going forward, we will sub-select reads with at least medium quality as defined by Bowtie2:
$ samtools view -h -b -q 20 mappings/evol1.sorted.dedup.bam > mappings/evol1.sorted.dedup.q20.bam
-h: Include the sam header-q 20: Only extract reads with mapping quality >= 20
Hint
I will repeat here a recommendation given at the source link above, as it is a good one: If you unsure what \(MAPQ\) scoring scheme is being used in your own data then you can plot out the \(MAPQ\) distribution in a BAM file using programs like the mentioned QualiMap or similar programs. This will at least show you the range and frequency with which different \(MAPQ\) values appear and may help identify a suitable threshold you may want to use.
Todo
Please repeat the whole process for the second evolved strain => mapping and post-processing.
Note
Should you be unable to process the second evolved strain look at the coding solutions here: Code: Mapping post-processing
5.10.3. Unmapped reads¶
We could decide to use Kraken2 like in section Taxonomic investigation to classify all unmapped sequence reads and identify the species they are coming from and test for contamination.
Lets see how we can get the unmapped portion of the reads from the bam-file:
$ samtools view -b -f 4 mappings/evol1.sorted.dedup.bam > mappings/evol1.sorted.unmapped.bam
# we are deleting the original to save space,
# however, in reality you might want to save it to investigate later
$ rm mappings/evol1.sorted.dedup.bam
# count the unmapped reads
$ samtools view -c mappings/evol1.sorted.unmapped.bam
-b: indicates that the output is BAM.-f INT: only include reads with this SAM flag set. You can also use the commandsamtools flagsto get an overview of the flags.-c: count the reads
Lets extract the fastq sequence of the unmapped reads for read1 and read2.
$ samtools fastq -1 mappings/evol1.sorted.unmapped.R1.fastq.gz -2 mappings/evol1.sorted.unmapped.R2.fastq.gz mappings/evol1.sorted.unmapped.bam
# delete not needed files
$ rm mappings/evol1.sorted.unmapped.bam
References
- LI2009
Li H, Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25 (14): 1754–1760.
- OKO2015
Okonechnikov K, Conesa A, García-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics (2015), 32, 2:292–294.