10. Variants-of-interest¶
10.1. Preface¶
In this section we will use our genome annotation of our reference and our genome variants in the evolved line to find variants that are interesting in terms of the observed biology.
Note
You will encounter some To-do sections at times. Write the solutions and answers into a text-file.
10.2. Overview¶
The part of the workflow we will work on in this section can be viewed in Fig. 10.1.
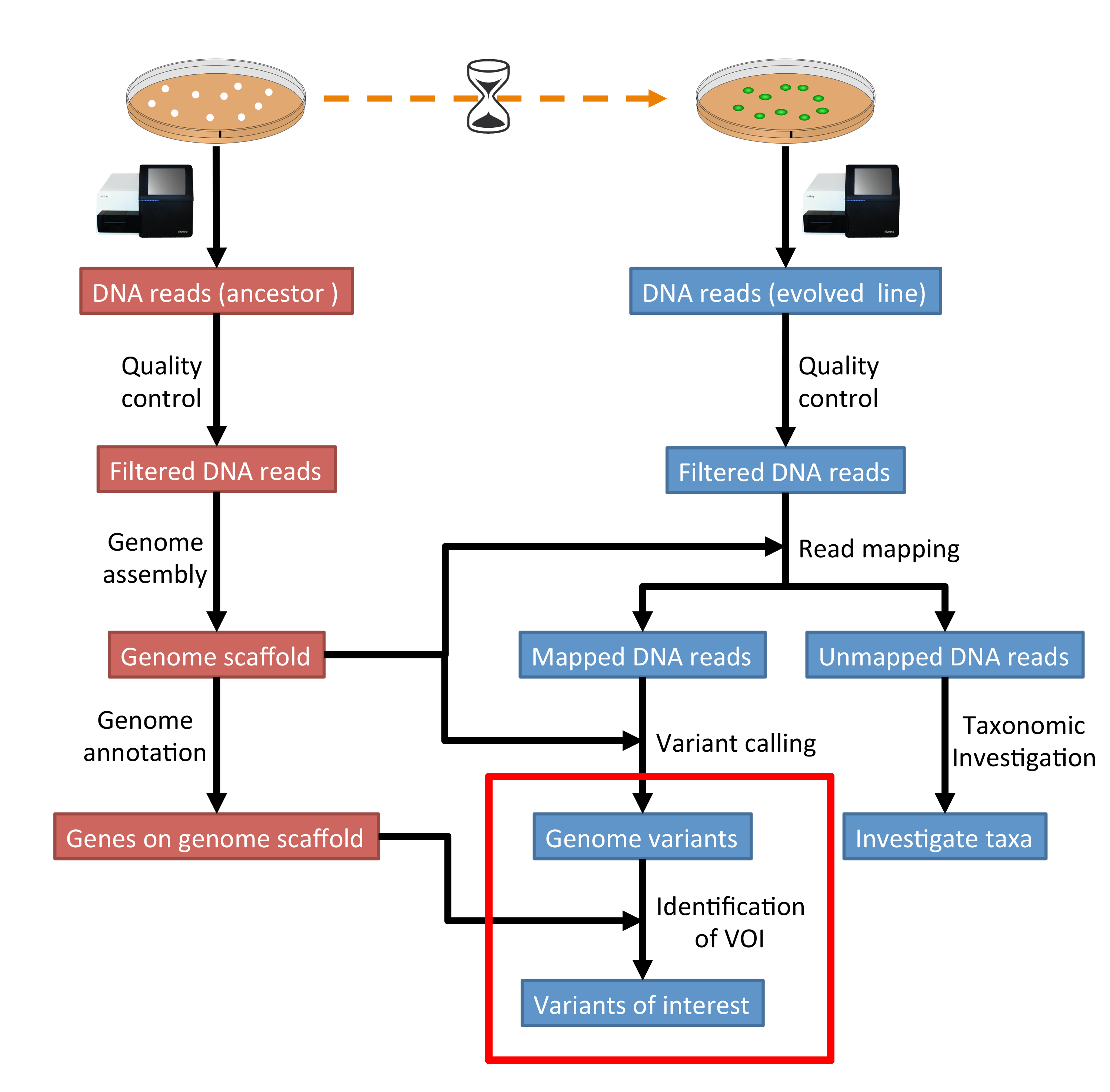
Fig. 10.1 The part of the workflow we will work on in this section marked in red.¶
10.3. Learning outcomes¶
After studying this section of the tutorial you should be able to:
Identify variants of interests.
Understand how the variants might affect the observed biology in the evolved line.
10.4. Before we start¶
Lets see how our directory structure looks so far:
$ cd ~/analysis
$ ls -1F
annotation/
assembly/
data/
kraken/
mappings/
phylogeny/
trimmed/
trimmed-fastqc/
variants/
Attention
If you have not run the previous sections on Genome assembly and Variant calling, you can download the variant calls and the genome assembly needed for this section here: Downloads. Download the files to the ~/analysis directory and decompress. Alternatively on the CLI try:
cd ~/analysis
wget -O assembly.tar.gz https://osf.io/t2zpm/download
tar xvzf assembly.tar.gz
eget -O variants.tar.gz https://osf.io/4nzrm/download
tar xvzf variants.tar.gz
10.5. General comments for identifying variants-of-interest¶
Things to consider when looking for variants-of-interest:
The quality score of the variant call.
Do we call the variant with a higher then normal score?
The mapping quality score.
How confident are we that the reads were mapped at the position correctly?
The location of the SNP.
SNPs in larger contigs are probably more interesting than in tiny contigs.
Does the SNP overlap a coding region in the genome annotation?
The type of SNP.
substitutions vs. indels
10.6. SnpEff¶
We will be using SnpEff to annotate our identified variants. The tool will tell us on to which genes we should focus further analyses.
10.6.1. Installing software¶
Tools we are going to use in this section and how to install them if you not have done it yet:
$ conda create -n voi snpeff genometools-genometools bedtools
Make a directory for the results (in your analysis directory) and change into the directory:
$ mkdir voi
# change into the directory
$ cd voi
10.6.2. Prepare SnpEff database¶
We need to create our own config-file for SnpEff.
Where is the snpEff.config:
$ find ~ -name snpEff.config
/home/guest/miniconda3/envs/voi/share/snpeff-4.3.1t-3/snpEff.config
This will give you the path to the snpEff.config.
It might be looking a bit different then the one shown here,
depending on the version of SnpEff that is installed.
Make a local copy of the snpEff.config and then edit
it with an editor of your choice:
$ cp /home/guest/miniconda3/envs/voi/share/snpeff-4.3.1t-3/snpEff.config .
$ nano snpEff.config
Make sure the data directory path in the snpEff.config looks like this:
data.dir = ./data/
There is a section with databases, which starts like this:
#-------------------------------------------------------------------------------
# Databases & Genomes
#
# One entry per genome version.
#
# For genome version 'ZZZ' the entries look like
# ZZZ.genome : Real name for ZZZ (e.g. 'Human')
# ZZZ.reference : [Optional] Comma separated list of URL to site/s Where information for building ZZZ database was extracted.
# ZZZ.chrName.codonTable : [Optional] Define codon table used for chromosome 'chrName' (Default: 'codon.Standard')
#
#-------------------------------------------------------------------------------
Add the following two lines in the database section underneath these header lines:
# my genome
mygenome.genome : EColiMut
Now, we need to create a local data folder called ./data/mygenome.
# create folders
$ mkdir -p ./data/mygenome
Copy our genome assembly to the newly created data folder.
The name needs to be sequences.fa or mygenome.fa:
$ cp ../assembly/scaffolds.fasta ./data/mygenome/sequences.fa
$ gzip ./data/mygenome/sequences.fa
Copy our genome annotation to the data folder.
The name needs to be genes.gff (or genes.gtf for gtf-files).
$ cp ../annotation/PROKKA_12345.gff ./data/mygenome/genes.gff
$ gzip ./data/mygenome/genes.gff
Now we can build a new SnpEff database:
$ snpEff build -c snpEff.config -gff3 -v mygenome > snpEff.stdout 2> snpEff.stderr
Note
Should this fail, due to gff-format of the annotation, we can try to convert the gff to gtf:
# using genometools
$ gt gff3_to_gtf ../annotation/PROKKA_12345.gff -o ./data/mygenome/genes.gtf
$ gzip ./data/mygenome/genes.gtf
Now, we can use the gtf annotation top build the database:
$ snpEff build -c snpEff.config -gtf22 -v mygenome > snpEff.stdout 2> snpEff.stderr
10.6.3. SNP annotation¶
Now we can use our new SnpEff database to annotate some variants, e.g.:
$ snpEff -c snpEff.config mygenome ../variants/evol1.freebayes.filtered.vcf > evol1.freebayes.filtered.anno.vcf
$ snpEff -c snpEff.config mygenome ../variants/evol2.freebayes.filtered.vcf > evol2.freebayes.filtered.anno.vcf
SnpEff adds ANN fields to the vcf-file entries that explain the effect of the variant.
Note
If you are unable to do the annotation, you can download an annotated vcf-file from Downloads.
10.6.4. Example¶
Lets look at one entry from the original vcf-file and the annotated one. We are only interested in the 8th column, which contains information regarding the variant. SnpEff will add fields here :
# evol2.freebayes.filtered.vcf (the original), column 8
AB=0;ABP=0;AC=1;AF=1;AN=1;AO=37;CIGAR=1X;DP=37;DPB=37;DPRA=0;EPP=10.1116;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=226.923;PAIRED=0.972973;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1155;QR=0;RO=0;RPL=12;RPP=12.9286;RPPR=0;RPR=25;RUN=1;SAF=26;SAP=16.2152;SAR=11;SRF=0;SRP=0;SRR=0;TYPE=snp
# evol2.freebayes.filtered.anno.vcf, column 8
AB=0;ABP=0;AC=1;AF=1;AN=1;AO=37;CIGAR=1X;DP=37;DPB=37;DPRA=0;EPP=10.1116;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=226.923;PAIRED=0.972973;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1155;QR=0;RO=0;RPL=12;RPP=12.9286;RPPR=0;RPR=25;RUN=1;SAF=26;SAP=16.2152;SAR=11;SRF=0;SRP=0;SRR=0;TYPE=snp;ANN=T|missense_variant|MODERATE|HGGMJBFA_02792|GENE_HGGMJBFA_02792|transcript|TRANSCRIPT_HGGMJBFA_02792|protein_coding|1/1|c.773G>A|p.Arg258His|773/1092|773/1092|258/363||WARNING_TRANSCRIPT_NO_START_CODON,T|upstream_gene_variant|MODIFIER|HGGMJBFA_02789|GENE_HGGMJBFA_02789|transcript|TRANSCRIPT_HGGMJBFA_02789|protein_coding||c.-4878G>A|||||4878|,T|upstream_gene_variant|MODIFIER|HGGMJBFA_02790|GENE_HGGMJBFA_02790|transcript|TRANSCRIPT_HGGMJBFA_02790|protein_coding||c.-3568G>A|||||3568|,T|upstream_gene_variant|MODIFIER|HGGMJBFA_02791|GENE_HGGMJBFA_02791|transcript|TRANSCRIPT_HGGMJBFA_02791|protein_coding||c.-442G>A|||||442|,T|upstream_gene_variant|MODIFIER|HGGMJBFA_02794|GENE_HGGMJBFA_02794|transcript|TRANSCRIPT_HGGMJBFA_02794|protein_coding||c.-1864C>T|||||1864|,T|upstream_gene_variant|MODIFIER|HGGMJBFA_02795|GENE_HGGMJBFA_02795|transcript|TRANSCRIPT_HGGMJBFA_02795|protein_coding||c.-3530C>T|||||3530|,T|upstream_gene_variant|MODIFIER|HGGMJBFA_02796|GENE_HGGMJBFA_02796|transcript|TRANSCRIPT_HGGMJBFA_02796|protein_coding||c.-4492C>T|||||4492|,T|downstream_gene_variant|MODIFIER|HGGMJBFA_02793|GENE_HGGMJBFA_02793|transcript|TRANSCRIPT_HGGMJBFA_02793|protein_coding||c.*840G>A|||||840|
When expecting the second entry, we find that
SnpEff added annotation information starting
with ANN=T|missense_variant|....
If we look a bit more closely we find that the variant
results in a amino acid change from a arginine to a
histidine (c.773G>A|p.Arg258His).
The codon for arginine is CGN and for histidine is
CAT/CAC, so the variant in the second nucleotide of
the codon made the amino acid change.
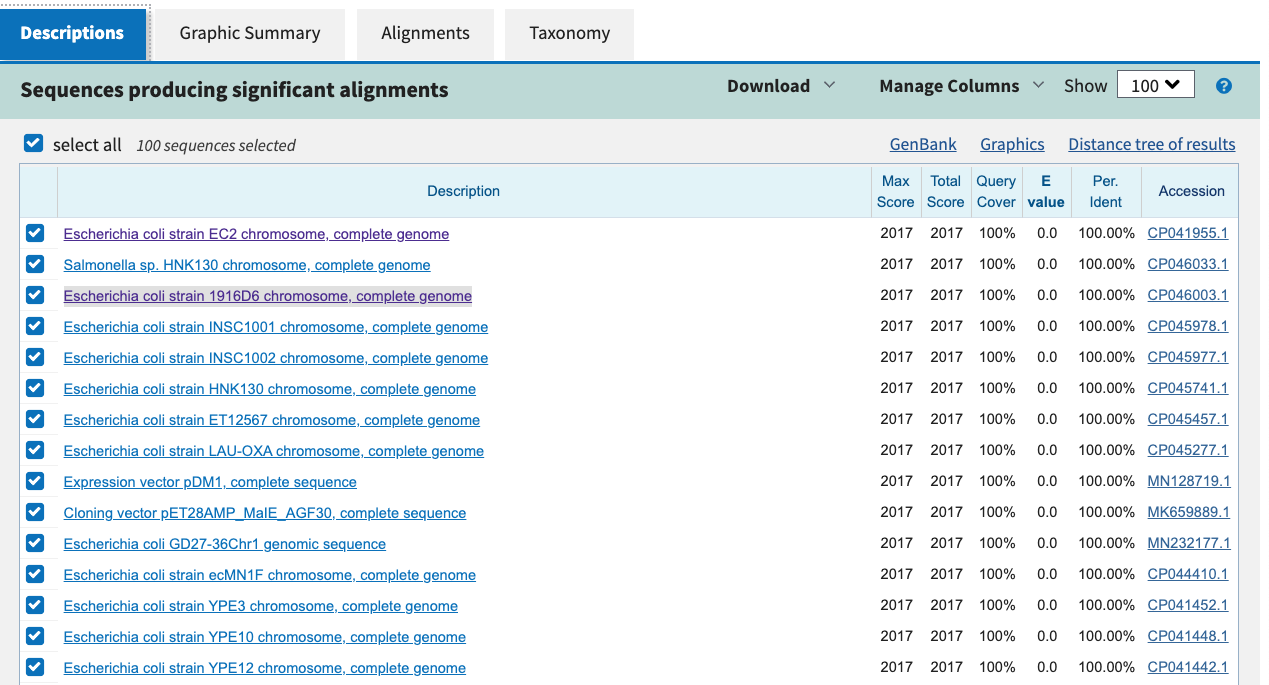
A quick BLAST search of the CDS sequence, where the variant
was found (extracted from the genes.gff.gz) shows that
the closest hit is a DNA-binding transcriptional regulator
from several different E.Coli strains.
# decompress annotation and genome
$ gzip -d data/mygenome/genes.gff.gz
$ gzip -d data/mygenome/sequences.fa.gz
# extract genes sequences
$ bedtools getfasta -fi data/mygenome/sequences.fa -bed data/mygenome/genes.gff > data/mygenome/genes.fa